We design and optimize enzymes using computational and experimental tools. We are interested in understanding the biophysical bases of enzyme function, uncovering the evolutionary implications of molecular recognition by enzymes, and engineering enzymes for applications aimed at addressing 21st-century challenges. These applications include biodegradation of pollutants and toxins, extending the range of enzyme-catalyzed chemistries to non-biological reactions, the design of enzymatic therapeutics, and making cancer chemotherapy more specific.
Enzymatic processes in nature are spatially organized. To develop the ability to similarly organize synthetic enzymatic pathways and develop efficient biosensors using enzymes (requiring high surface:volume), we have developed a modular design approach that allows construction of supramolecular assemblies in response to chemical and/or optical stimuli (Yang et al. ChemBioChem 2017 ). More recently, we have built fractal supramolecular topologies with extremely high surface area:volume ratios that organize component enzymes into hyperbranched dendritic supramolecular topologies (Hernandez, Hansen et al. submitted). Although fractals are ubiquitous in nature, our studies represent the first instance of designing such topologies using protein self-assembly. These studies are in collaboration with structural biologists Wei Dai, Sanghyuk Lee (Rutgers) and enzyme biochemist Lawrence Wackett (Minnesota), and aimed at enhancing the efficiency of pollutant biosensing and biodegradation.
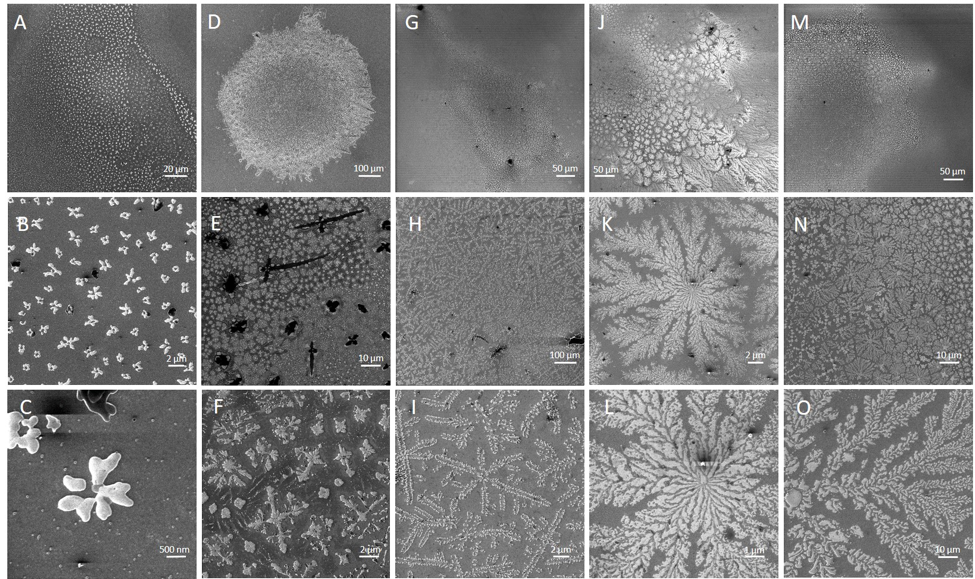
Helium Ion microscopy images of designed fractal topologies
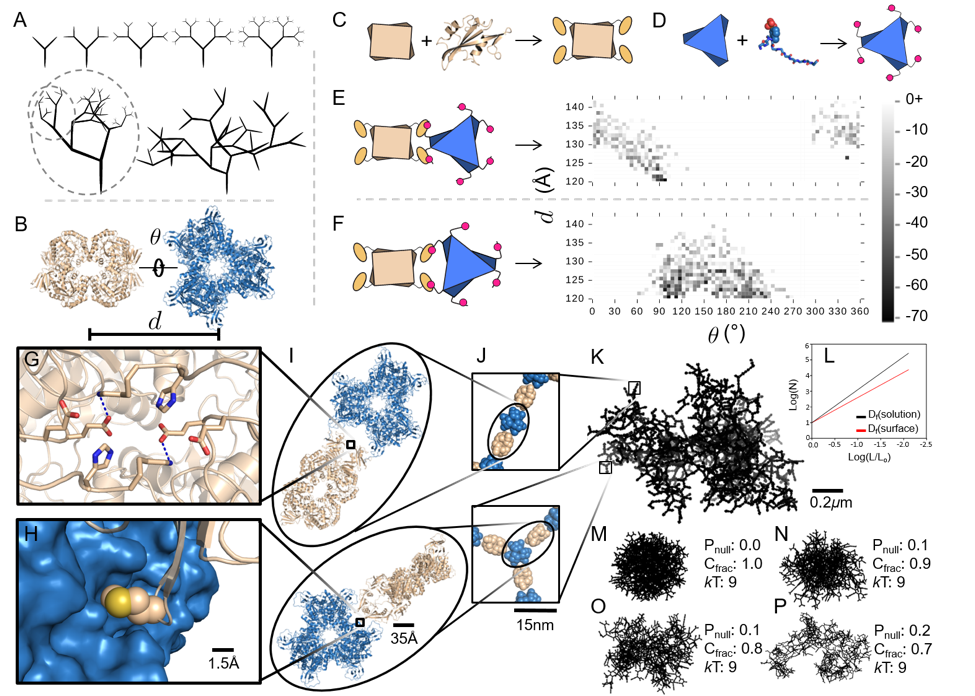
Computational design approach
Site-selective proteolytic cleavage is a ubiquitous post-translational modification involved in the transfer of biological information (e.g., via cascades) in many cellular processes and their dysfunction. Proteases with “dialed in” substrate selectivities would be ideal catalytic drugs designed to irreversibly neutralize their target substrates (e.g., viral coat proteins) if their substrate selectivity can be precisely controlled. No robust and general method is available for protease substrate specificity design, in spite of ~25 years of efforts by protein chemists and chemical engineers. Our approach is to develop a mechanism-guided biophysical framework that allows design for both positive and negative substrate specificity, and tightly couple it with high-throughput experimental testing. We have developed a new computational design approach (Rubenstein et al. PLOS Comp. Biol, 2017) combining Rosetta and Amber calculations (Pethe et al. J. Mol. Biol. 2017), and developing high-throughput characterization and supervised learning approaches that combine computational predictions with experimental data obtained using yeast-based screening and deep sequencing (Pethe, Rubenstein et al. submitted). Our approach allows simultaneously querying and identifying millions of peptide sequences for cleavability.
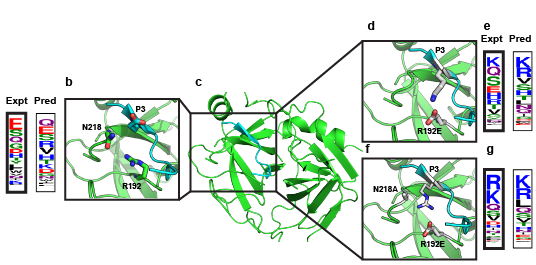
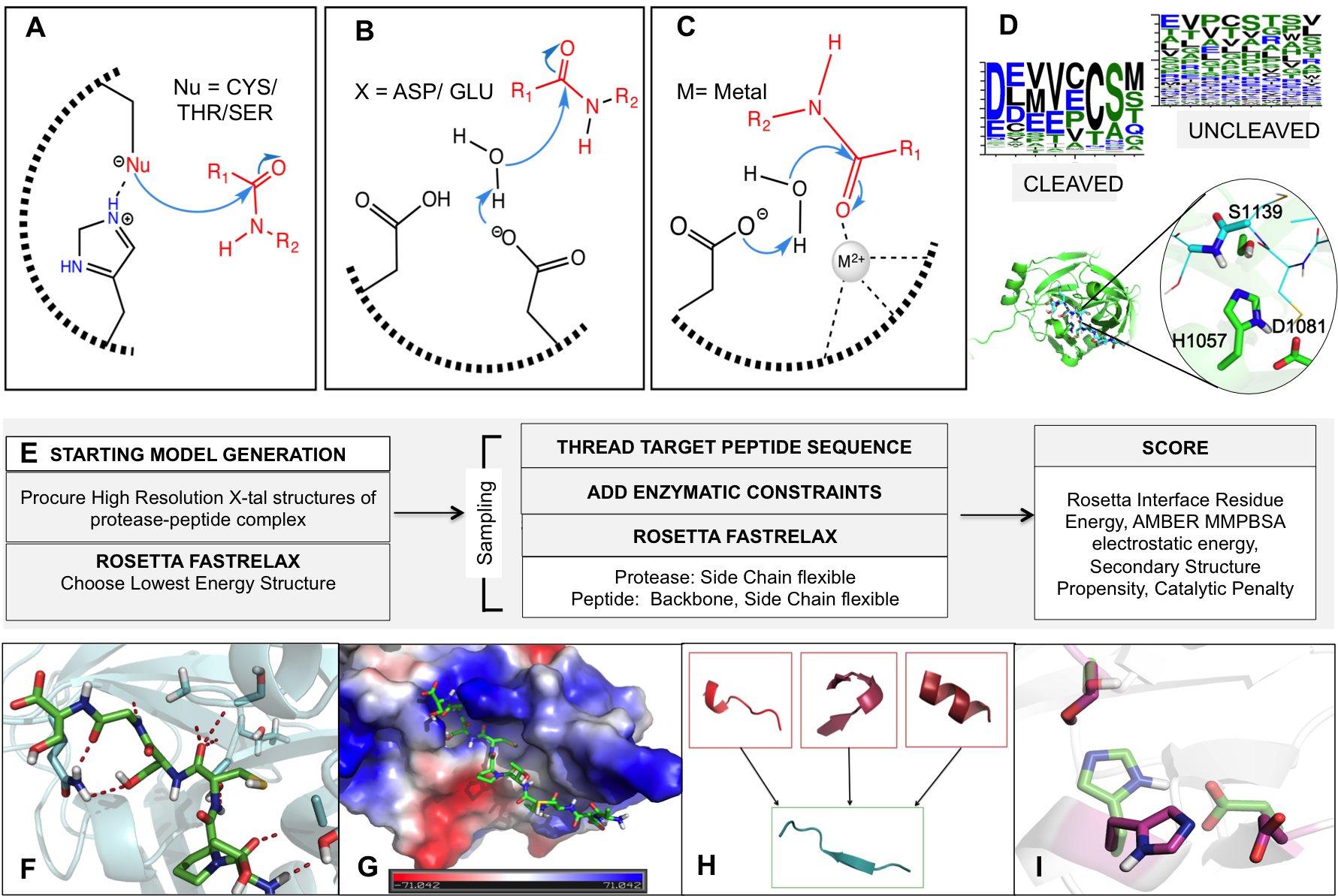
Prediction of specificity profile changes upon mutation (Rubenstein et al. PLOS Comp Biol) and scheme for scoring protease-substrate complexes (Pethe et al. JMB)
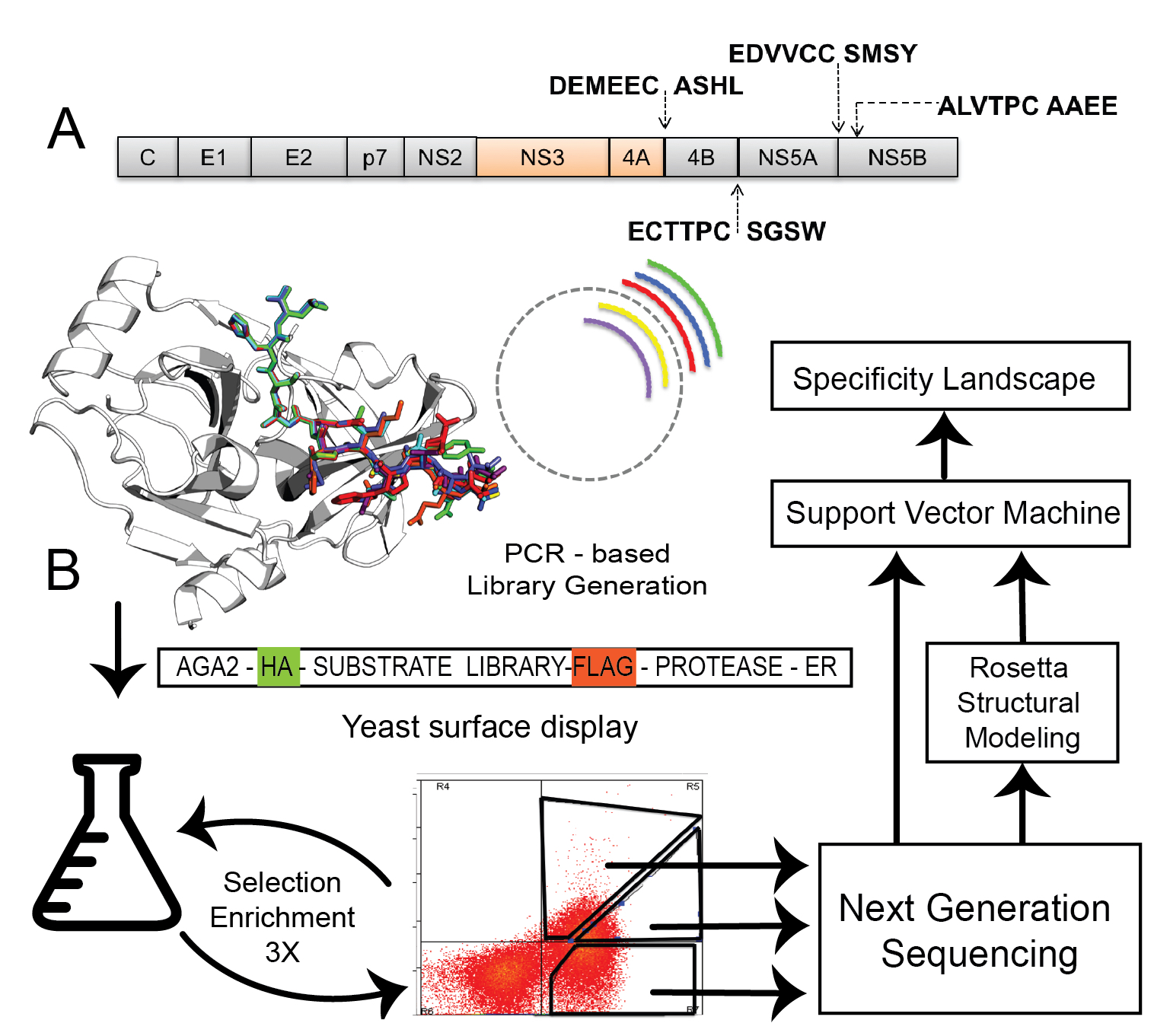
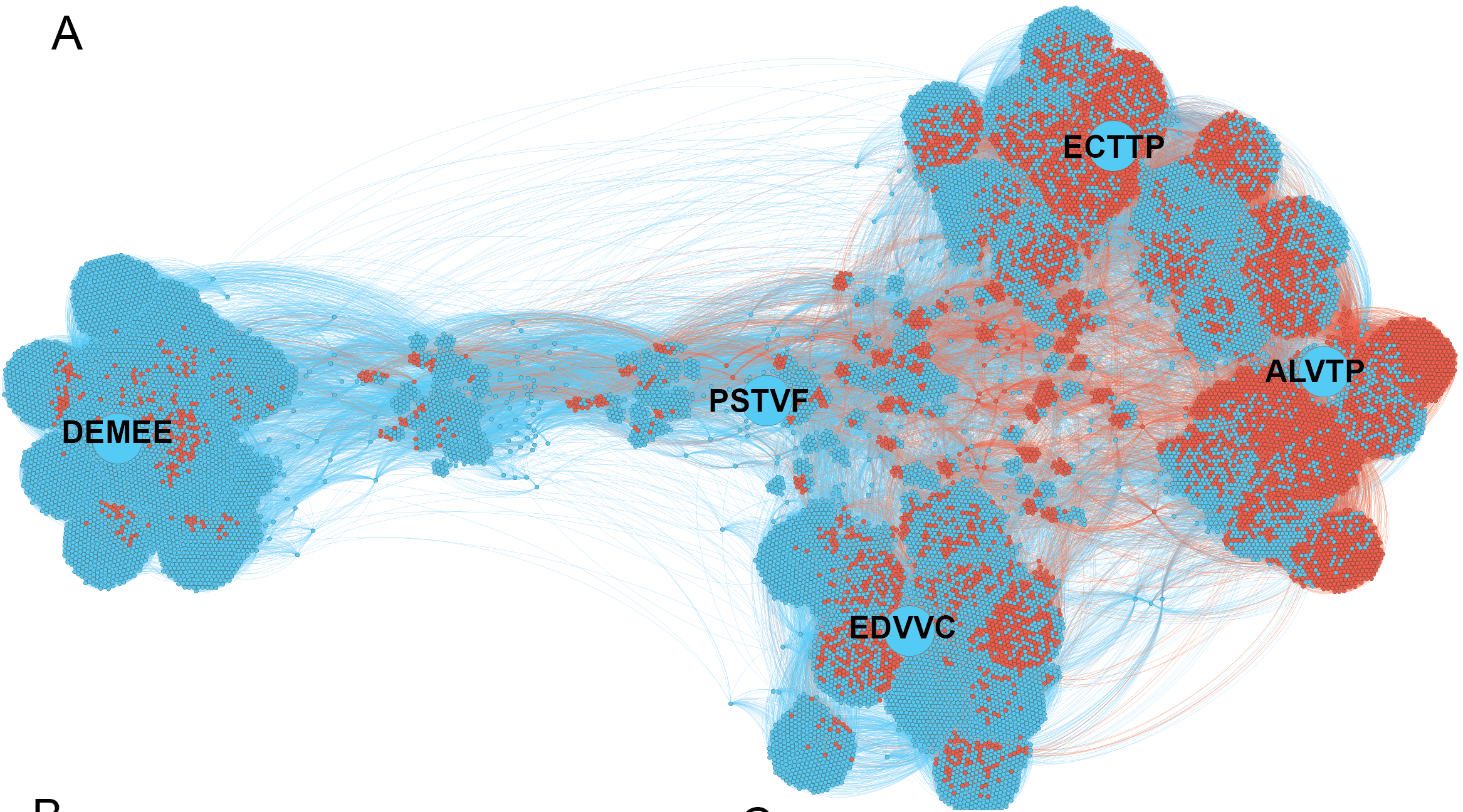
Supervised learning of a specificity landscape of a protease by combining yeast surface display, molecular simulations and machine learning (Pethe, Rubenstein et al. submitted)
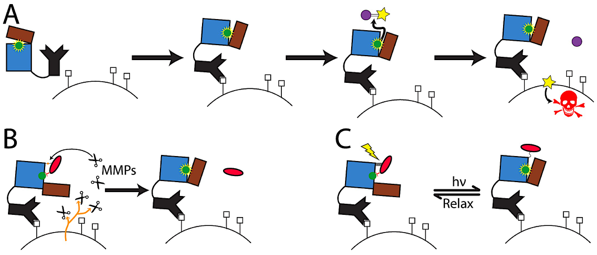
We are developing the ability to design enzymes such that they can be activated by environmental stimuli or external triggers such as light. The application we are pursuing is Directed Enzyme Prodrug Therapy, which is a promising approach to attenuate side-effects and thereby increase the therapeutic efficacy of conventional chemotherapy. In this approach an exogenous enzyme, targeted to the tumor by, for example, an antibody, activates a prodrug to generate toxicity locally. Animal studies and clinical trials have shown that slow clearance leading to activity of enzyme in non-tumor tissue is a major limitation. Computationally designed “smart” enzymes, that are constitutively inactive but are activatable by a tumor-specific stimulus (MMP-2 protease) or light (via use of attached azobenzene dyes) are expected to overcome these limitations. We have obtained good starting leads straight from computational design (~10X switches) (Blacklock et al. JACS 2018), developed high-throughput screening approaches (Yachnin & Khare, Prot. Eng. Des. Sel. 2017). Biological characterization of designed enzymes is ongoing (Justin Drake, Rutgers).
Enzymes are the workhorses of biology, and greatly important tools with many applications in biotechnology and medicine. The marginal stability of most natural proteins (-5-15 kcal/mol) presents a major challenge for the exploitation of the amazing properties of natural and engineered enzymes. For example, in biocatalytic processes, e.g., synthesis of drugs, fine chemicals and biofuels, enzymes are subject to harsh operational conditions such as high temperatures and presence of organic co-solvents (e.g., methanol, DMSO etc.), so more stable enzymes can withstand these demanding conditions better. More stable enzymes are also more “evolvable” i.e., they can better tolerate chemical changes that impart them new or improved functions. Enzyme stabilization represents a crucial, and often obligatory step, for the utilization of their attractive properties. Currently available methods for protein stabilization, such as directed evolution and consensus mutagenesis, can be time- and labor-intensive and often involve extensive amino acid substitutions, which may impair the activity and/or selectivity of the enzyme. We have developed a computational design method (Rosetta-guided protein stapling, R-GPS) for enzyme stabilization that uses structure-based modeling to introduce covalent ‟staples” in a protein scaffold via genetically encodable noncanonical amino acids. This method was applied to obtain stapled variants of a stereoselective cyclopropanation biocatalyst featuring greatly increased thermostability and robustness to high concentrations of organic cosolvents (Moore et al. PNAS 2017). In collaboration with Fasan (Rochester) and Ando (Princeton) labs, and using iterative design-build-test-characterize cycles, we are now extending this minimally invasive strategy for protein stabilization to (1) a variety of other enzymes and proteins, and (2) to genetically encodable non-canonical amino acids that enable other kinds of crosslinking chemistries.

We remain interested in the development of new computational design methods and refinement of existing methodology, as these are the foundations of our research program. Our efforts include introducing a new (Potts-model-based) modeling/design algorithm in Rosetta (Rubenstein et al. PLOS Comp. Biology. 2017), a method for protein photosensitization using chemically crosslinked small molecules (Blacklock et al., JACS 2018), and a method for designing nested (domain-inserted) proteins (Blacklock et al. Proteins 2018). We have developed new algorithms for multinuclear metalloprotein design (Hansen & Khare, Prot. Sci., 2017) and are testing them with Vikas Nanda’s lab (Rutgers; Nanda et al. BBRC 2016). We are pursuing force-field improvements in Rosetta, working on a project with David Case (Rutgers) (Rubenstein et al JCTC 2018) to compare and productively combine energy evaluations using Rosetta and Amber approaches.